

Estrutura de glicogênio, síntese, degradação, funções
- 4804
- 134
- Dennis Heidenreich
Ele Glicogênio É o carboidrato de armazenamento da maioria dos mamíferos. Os carboidratos são comumente chamados de açúcares e são classificados de acordo com o número de resíduos causados por hidrólise (monossacarídeos, dissacarídeos, oligossacarídeos e polissacarídeos)))).
Os monossacarídeos são os carboidratos mais simples que são classificados de acordo com o número de carbonos contidos em sua estrutura. Existem então as triosas (3C), tetrosas (4c), pentosas (5c), hexósio (6c), heptosase (7c) e octo (8c).
Estrutura de glicogênio químico mostrando ligações glicosídicas (Fonte: Glykogen.SVG: Trabalho derivado de neurotoger: Marek M [Domínio Público] via Wikimedia Commons) De acordo com a presença do grupo aldeído ou do grupo Cetona, esses monossacarídeos também são classificados como aldies ou cetosas, respectivamente,.
Os dissacarídeos dão origem à hidrólise, dois monossacarídeos simples, enquanto os oligossacarídeos produzem 2 a 10 unidades de monossacarídeos e polissacarídeos produzem mais de 10 monossacarídeos.
O glicogênio é, do ponto de vista bioquímico, um polissacarídeo composto por cadeias ramificadas de uma aldose de seis carbonos, ou seja, uma hexose conhecida como glicose. Graficamente ele pode ser representado para glicogênio como uma árvore de glicose. Isso também é chamado de amido animal.
A glicose nas plantas é armazenada como amido e em animais como glicogênio, que é armazenado principalmente no fígado e no tecido muscular.
No fígado, o glicogênio pode estabelecer 10% de sua massa e 1% da massa muscular. Como em um homem de 70 kg, o fígado pesa cerca de 1800 g e os músculos cerca de 35 kg, a quantidade total de glicogênio muscular é muito maior que o hepático.
[TOC]
Estrutura
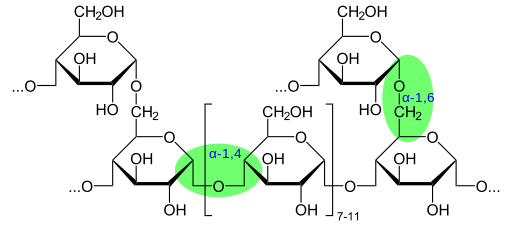
O peso molecular do glicogênio pode atingir 108 g/mol, equivalente a 6 × 105 moléculas de glicose. O glicogênio é composto de múltiplas cadeias ramificadas α-d-glicrose. A glicose (C6H12O6) é uma aldohexosa que pode ser representada de maneira linear ou cíclica.
O glicogênio possui uma estrutura muito ramificada e compacta com cadeias de 12 a 14 resíduos de glicose na forma de α-d-glicose que estão ligados a ligações glucosídicas α- (1 → 4). As ramificações da cadeia são formadas por links glucosídicos α- (1 → 6).
O glicogênio, como o amido que é ingerido na dieta, fornece a maioria dos carboidratos que o corpo precisa. No intestino, esses polissacarídeos são degradados por hidrólise e depois absorvidos em direção à torrente circulatória principalmente como glicose.
Três enzimas: ß-amilase, α-amilase e amil-α- (1 → 6) -glucosidase são responsáveis pela degradação intestinal de glicogênio e amido.
A α-amilase hidrolisa aleatoriamente as ligações α-(1 → 4) das cadeias laterais de glicogênio e amido e, portanto, recebe o nome da endoglysidase. A ß-amyla é uma exoglicosidase que está liberando os links glicosídicos de α- (1 → 4) da α- (1 → 4) das extremidades das cadeias mais externas sem atingir as ramificações.
Em vista do fato de que nem a ß-amilase nem a α-amilase degradam ramos, o produto final de sua ação é uma estrutura altamente ramificada de cerca de 35 a 40 resíduos de glicose que são chamados de dextrina limite.
A dextrina limite é finalmente hidrolisada nos pontos de ramificação que possuem ligações α- (1 → 6) através das amyle-α- (1 → 6) -glucosidase, também conhecida como enzima "difamatória". As cadeias liberadas por este defloat são degradadas por ß-amilase e α-amilase.
Quando o glicogênio ingerido entra como glicose, aquele encontrado nos tecidos deve ser sintetizado pelo organismo da glicose.
Pode atendê -lo: purinas: características, estrutura, funçõesSíntese
A síntese de glicogênio é chamada de glicogênese e ocorre especialmente no músculo e no fígado. A glicose que entra no organismo com a dieta passa para a torrente circulatória e de lá dentro das células, onde é imediatamente fosforilada por uma enzima chamada glicoquinase.
Glucoquinase fosforyil para glicose em carbono 6. O ATP fornece fósforo e energia para esta reação. Como resultado, a glicose 6-fosfato é formada e um ADP é liberado. Então, a glicose 6-fosfato se torna glicose 1-fosfato pela ação de uma fosfoglucomutase que muda o fósforo da posição 6 para a posição 1.
A glicose de 1-fosfato é ativada para a síntese de glicogênio, que implica a participação de um conjunto de outras três enzimas: a pirofosforlase UDP-glicose, glicogênio sintético e amilo- (1,4 → 1.6) -glicosiltransferase.
A glicose-1-fosfato, juntamente com a uridina trifosfato (UTP, um nucleosídeo do trifosfato de uridina) e pela ação da UDP-glicose-pirofosforilase, formam o complexo de uridina difosfato-glicose (UDP GLC) (UDP GLC). No processo, um íon pirofosfato é hidrolisado.
Em seguida, a enzima glicogênio sintetizada forma uma ligação glucosídica entre o C1 do complexo UDP GLC e o C4 de um resíduo terminal de glicose de glicogênio, e o UDP UDP é liberado do complexo de glicose ativado por UDP. Para que essa reação ocorra, deve haver uma molécula de glicogênio pré -existente chamada "glicogênio primário".
O glicogênio primordial é sintetizado em uma proteína de priming, a glicogenina, que possui 37 kDa e glysila em um resíduo de tirosina usando o complexo UDP GLC. A partir daí, eles são links α-d-glicose resíduos com 1 → 4 links e uma pequena cadeia é formada na qual os atos de glicogênio da sintetização.
Uma vez que a cadeia inicial liga pelo menos 11 resíduos de glicose, a enzima ramificada ou a amile (1,4 → 1,6) -glicosiltransferase transfere uma peça de corrente de 6 ou 7 resíduos de glicose para a cadeia adjacente na posição 1 → 6, que estabelece um ramo de um ramo apontar. A molécula de glicogênio assim construída está crescendo por acréscimos de unidades de glicose com links glicosídicos 1 → 4 e mais ramificações.
Degradação
A degradação do glicogênio é chamada de glicogenólise e não é equivalente ao caminho reverso de sua síntese. A velocidade desta rota é limitada pela velocidade da reação catalisada pela fosforilase glicogênio.
O glicogênio da fosforlase é responsável pela divisão (fosforólise) dos links 1 → 4 das cadeias de glicogênio, liberando glicose 1-fosfato. A ação enzimática começa nas extremidades das correntes mais externas e é removida sequencialmente até que 4 resíduos de glicose permaneçam em cada lado das ramificações.
Então, outra enzima, a α- (1 → 4) → α- (1 → 4) glucano transferas, deixa o ponto de ramificação exposto ao transferir uma unidade de trissacarídeo de um ramo para outro. Isso permite que a amilo- (1 → 6) -glucosidase (enzima urbana) hidrolys. A ação combinada dessas enzimas termina completamente se dividindo em glicogênio.
Como a reação inicial da fosfomutase é reversível, a glicose de 6-fosfato pode ser formada a partir de resíduos de glicose 1-fosfato divididos de glicogênio. No fígado e rim, mas não no músculo, há uma enzima, glicose-6-fosfatase, capaz de se reunir com glicose de 6 fosfato e transformá-la em glicose livre.
Pode atendê -lo: FotóliseA glicose emformorilada pode se espalhar para o sangue, e é assim que a glicogenólise hepática se reflete em um aumento nos valores de glicose no sangue (glicemia).
Regulação da síntese e degradação
De síntese
Esse processo é exercido em duas enzimas fundamentais: glicogênio sintetSase e glicogênio fosforilase, de modo que, quando um deles é ativado, o outro está em seu estado inativo. Este regulamento impede que as reações opostas de síntese e degradação ocorram simultaneamente.
A forma ativa e a forma inativa de ambas as enzimas são muito diferentes, e a interconversão das formas ativas e inativas de fosforilase e glicogênio sintético está sujeito a controle hormonal rigoroso.
A adrenalina é um hormônio liberado da medula adrenal, e o glucagon é outro que ocorre na parte endócrina do pâncreas. O pâncreas endócrino produz insulina e glucagon. Langerhans Islets α são os que sintetizam o glucagon.
Adrenalina e glucagon são dois hormônios liberados quando a energia é necessária em resposta à diminuição dos níveis de glicose no sangue. Esses hormônios estimulam a ativação do glicogênio da fosforilase e inibem a glicogênio da sintetase, estimulando a glicogenólise e inibindo a glicogênese.
Enquanto a adrenalina exerce sua ação sobre músculos e fígado, o glucagon age apenas no fígado. Esses hormônios são unidos a receptores membranais específicos na célula branca, que ativa o Cyclasa Adenilate.
A ativação do adenilato da ciclase inicia uma cascata enzimática que, por um lado, ativa uma proteinquinase dependente de AMPC que inativa a glicogênio sintético e ativa a fosforilase de glicogênio por fosforilação (direta e indiretamente, respectivamente).
O músculo esquelético tem outro mecanismo para a ativação do glicogênio da fosforilase através do cálcio, que é liberado como conseqüência da despolarização da membrana muscular no início da contração.
De degradação
As cachoeiras enzimáticas descritas acima acabam aumentando os níveis de glicose e, quando atingem um certo nível, a glicogênese é ativada e a glucogenólise é inibida, também inibindo a liberação adicional de adrenalina e glucagon.
A glicogênese é ativada pela ativação da fosfatase fosforilase, uma enzima que regula a síntese de glicogênio por vários mecanismos, o que implicam a inativação do inibidor da quinase fosforlase e fosforilase α, que é uma glicogênio da sintetase.
A insulina promove a entrada de glicose em células musculares, aumentando os níveis de glicose de 6-fosfato, o que estimula a defosforilação e a ativação do glicogênio da sintetSase. Assim, a síntese começa e a degradação do glicogênio é inibida.
Funções
O glicogênio muscular constitui uma reserva de energia para o músculo que, como as gorduras de reserva, permite que o músculo cumpra suas funções. Sendo uma fonte de glicose, o glicogênio muscular é usado durante o exercício. Essas reservas aumentam com o treinamento físico.
No fígado, o glicogênio também constitui uma importante fonte de reserva para as funções do órgão e para a contribuição da glicose para o resto do corpo.
Esta função do glicogênio hepático se deve ao fato de o fígado contém glicose 6-fosfatase, uma enzima capaz de eliminar o grupo fosfato de glicose de 6-fosfato e transformá-lo em glicose livre. A glicose livre, diferentemente da glicose fosforilada, pode ser espalhada pela membrana dos hepatócitos (células hepáticas).
Pode atendê -lo: esporulação: em plantas, em fungos e em bactériasÉ assim que o fígado pode fornecer glicose à circulação e manter níveis estáveis de glicose, mesmo em condições prolongadas de jejum.
Essa função é de grande importância, uma vez que o cérebro é nutrido quase exclusivamente da glicose no sangue, hipoglicemia tão grave (concentrações de glicose no sangue muito baixas) podem causar perda de conhecimento.
Doenças relacionadas
Doenças relacionadas a glicogênio recebem o nome genérico de "doenças de armazenamento de glicogênio".
Essas doenças constituem um grupo de patologias hereditárias caracterizadas pelo depósito nos tecidos de quantidades anormais ou tipos de glicogênio.
A maioria das doenças de armazenamento de glicogênio é causada por um déficit da natureza genética de qualquer uma das enzimas envolvidas no metabolismo do glicogênio.
Eles são classificados em oito tipos, a maioria dos quais tem seus próprios nomes e cada um deles é produzido por um déficit enzimático diferente. Alguns são mortais em estágios muito iniciais da vida, enquanto outros são acompanhados pela fraqueza e déficit muscular durante o exercício.
Exemplos excelentes
Algumas das doenças relacionadas a glicogênio mais proeminentes são as seguintes:
- A doença de Von Gierke ou a doença de armazenamento de glicogênio do tipo I é produzida por um déficit de glicose 6-fosfatase no fígado e rim.
É caracterizada pelo crescimento hepático anormal (hepatomegalia) devido ao acúmulo exagerado de glicogênio e hipoglicemia, uma vez que o fígado se torna incapaz de fornecer glicose à circulação. Pacientes com esta condição têm alterações de crescimento.
- A doença de Pompe ou tipo II é devida a um déficit α-déficit (1 → 4) -glucano 6-glicosiltransferas no fígado, coração e músculos esqueléticos. Esta doença, como Andersen ou Tipo IV, é letal antes dos dois anos de vida.
- A doença de McArdle ou Tipo V tem um déficit muscular da fosforilase e é acompanhada pela fraqueza muscular, diminuição da tolerância ao exercício, acúmulo anormal do glicogênio muscular e ausência de lactato durante o exercício durante o exercício.
Referências
- Bhattacharya, k. (2015). Investigação e gerenciamento das doenças de armazenamento de glicogênio hepático. Pediatria de tradução, 4(3), 240-248.
- Dagli, a., Sentner, c., & Weinstein, D. (2016). Doença de armazenamento de glicogênio tipo III. Revisões de genes, 1-16.
- Guyton, a., & Hall, J. (2006). Livro de fisiologia médica (11ª ed.). Elsevier inc.
- Mathews, c., Van holde, k., & Ahern, k. (2000). Bioquímica (3ª ed.). São Francisco, Califórnia: Pearson.
- McKiernan, p. (2017). Patobiologia do desejo hepático de armazenamento de glicogênio. Curr Pathobiol Rep.
- Murray, r., Bender, d., Botham, k., Kennelly, p., Rodwell, v., & Weil, P. (2009). Bioquímica ilustrada de Harper (28ª ed.). McGraw-Hill Medical.
- Nelson, d. eu., & Cox, M. M. (2009). Lehninger Principles of Biochemistry. Edições Omega (5ª ed.).
- Rawn, j. D. (1998). Bioquímica. Burlington, Massachusetts: Neil Patterson Publishers.
- Tarnopolsky, m. PARA. (2018). Miopatias relacionadas a distúrbios do metabolismo do glicogênio. Neuroterapêuticos.
- « História de argônio, estrutura, propriedades, usa
- Função bijetiva O que é, como é feito, exemplos, exercícios »